SELTENE ERKRANKUNGEN
Hereditäre Tyrosinämie Typ 1
Bei der Tyrosinämie Typ 1 (HT-1) handelt es sich um einen angeborenen Defekt im Tyrosin-Abbaustoffwechsel, dem eine gestörte Funktion der Fumarylacetoacetathydrolase (FAH) zugrunde liegt. Diese Erkrankung ist durch eine fortschreitende Lebererkrankung, Störung der tubulären Nierenfunktion und Porphyrie-ähnliche Beschwerden gekennzeichnet.
Geschichte der Erkrankung und ihrer medikamentösen Behandlung

Die US-amerikanische Biochemikerin Grace Medes (09.11.1886 – 31.12.1967) kreiert den Begriff Tyrosinosis [Tyrosinose] bei ihrer Beschreibung der ersten biochemischen Befunde bei einem 49-jährigen Mann mit Myasthenia gravis. In der Abhandlung mit dem Titel „A new error of tyrosine metabolism: Tyrosinosis“ [Ein neuer Defekt im Tyrosinstoffwechsel: Tyrosinose] berichtet sie über eine ungewöhnliche Reduktionssubstanz im Harn dieses Patienten, die als 4-Hydroxyphenylpyruvat (4-HPP) identifiziert wird, und ordnet den Defekt bei der Tyrosinose einem Mangel an 4-Hydroxyphenylpyruvat-Dioxygenase (4-HPPD) zu. Diese Erkrankung wurde in den Genkatalog „Mendelian Inheritance in Man“ mit Code 76800 aufgenommen.
Grace Medes wird die Garvan Medaille verliehen, eine Auszeichnung für besondere Leistungen von Wissenschaftlerinnen auf dem Gebiet der Chemie.
Sakai und Kitagawa berichten über den Fall eines zweijährigen Jungen mit ausgeprägter Hepatosplenomegalie und Gedeihstörung. Eine Urinuntersuchung ergibt eine große Menge an 4-Hydroxyphenyllactat (4-HPL) und eine kleine Menge an 4-HPP, 4-Hydroxyphenylacetat (4-HPA) und Tyrosin. Die physikalischen Befunde und biochemischen Beobachtungen lassen die Autoren daran denken, dass sich der bei dem Patienten vorliegende angeborene Defekt im Tyrosinstoffwechsel von dem unterscheidet, über den Medes ein Viertel Jahrhundert zuvor berichtet hatte. Sie veröffentlichten ihre erste Abhandlung über diesen Fall unter dem Titel „An atypical case of tyrosinosis“ [Ein untypischer Fall von Tyrosinose].
Dritte Abhandlung von Sakai, Kitagawa und Yoshioka, in der sie die Beobachtungen zur Pathologie und Biochemie der Organgewebe dieses Patienten beschreiben.
Prof. Sverre Halvorsen (24.07.1925 – 08.08.2012) und Leiv R. Gjessing zeigen erstmalig die überaus wichtige Rolle einer Phenylalanin- und Tyrosin-armen Diät bei der Pathogenese tubulärer Nierenschäden bei einem 2-jährigen Mädchen auf.
Prof. Sverre Halvorsen und Leiv R. Gjessing organisieren ein Symposium zur Tyrosinose zu Ehren von Grace Medes. Es ist hervorzuheben, dass Dr. Asbjorn Fölling, emeritierter Professor an der Universität Oslo, der 1934 den ersten Fall einer Phenylketonurie beschrieben hat, an der Konferenz teilnahm.
Dr. Jean Larochelle macht den Vorschlag, die Erkrankung Hereditäre Tyrosinämie zu nennen.
Das Enzym Fumarylacetoacetathydrolase (FAH) wird als das fehlerhafte Enzym identifiziert, das zu HT-1 führt. FAH ist das letzte Enzym im katabolen Stoffwechselweg von Tyrosin.
Dr. Richard Gagné beschreibt eine HT-1-Nachweismethode durch Messung des Succinylaceton-Spiegels im Fruchtwasser, die ab der 16. Schwangerschaftswoche eingesetzt werden kann.
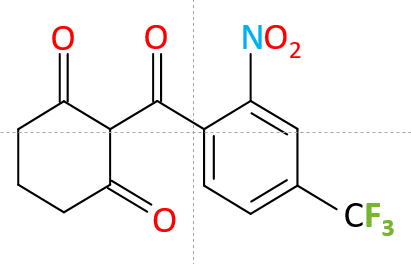
Zeneca Agrochemicals entdeckt das Herbizid Nitisinon aus der Klasse der Triketone. Es ist ein Derivat von Leptospermon, das vom Flaschenbürstengras gebildet wird. Nitisinon ist ein Hemmstoff der 4-Hydroxyphenylpyruvat-Dioxygenase (HPPD). HPPD ist das zweite von fünf Enzymen, die am katabolen Stoffwechselweg von Tyrosin beteiligt sind, und FAH ist das letzte Enzym. Es ist hervorzuheben, dass 4 der 5 Enzyme mit angeborenen Stoffwechseldefekten verbunden sind (Tyrosinämie Typ 2 und 3, Hawkinsinurie, Alkaptonurie und Hyperphenylalaninämie aufgrund Dehydratase-Mangel).
Tyrosinämie, Typ 1 (HT-1) wird in die Datenbank OMIM unter Code 276700 aufgenommen.
Ein schwerkrankes, zwei Monate altes Baby ist der erste erfolgreich mit Nitisinon wegen HT-1 behandelte Patient in einer klinischen Studie, die von der Schwedischen Arzneimittel-Agentur im Februar desselben Jahres genehmigt wurde. Daraufhin wurden weitere HT-1-Patienten mit Nitisinon im Rahmen eines Härtefall-Programms behandelt.

Sven Lindstedt (1927-2015) und Prof. Elisabeth Holmes (1947-2015) berichten, dass HT-1-bedingte neurologische Schübe durch die Anwendung von Nitisinon in Kombination mit einer angemessenen Diät gebessert und verhindert werden. Diese Entdeckung gab Patienten neue Hoffnung, denen sonst nur eine Transplantation geblieben wäre.
Orfadin®, das erste Arzneimittel gegen HT-1 mit dem Wirkstoff Nitisinon, wird in den USA von der FDA zugelassen. Aufgrund seiner Instabilität bei Raumtemperatur muss Orfadin® bei 2-8 °C gelagert werden.
Orfadin® wird von der EMA in Europa zugelassen.
Eine Arbeitsgruppe aus europäischen und kanadischen HT-1-Spezialisten veröffentlicht die ersten Empfehlungen für die HT-1-Therapie.
Symptomatik
Die typischen klinischen Symptome setzen vor dem 2. Lebensjahr ein, wobei bei den meisten Kindern Hinweise auf akutes Leberversagen und Nierenfunktionsstörungen vor dem 6. Lebensmonat auftreten. Neurologische Schübe in Form schmerzhafter Episoden, die Gliedmaßen und/oder die Funktion der Bauchorgane betreffen und mit Hypertonie und Hyponatriämie einhergehen, können jederzeit auftreten und zu respiratorischer Insuffizienz und Tod führen. Bei einigen erkrankten Kindern können ab dem 2. Lebensjahr vereinzelte Koagulopathien oder andere Anzeichen einer Leberinsuffizienz, eine tubuläre Nierenerkrankung, hypophosphatämische Rachitis und Gedeihstörungen auftreten. Bei allen Kindern mit HT-1 ist das Risiko für hepatozelluläre Karzinome stark erhöht.
Inzidenz
1 von 100.000-120.000 Menschen. Dieser Typ der Tyrosinämie tritt sehr viel häufiger in der kanadischen Provinz Quebec und in den skandinavischen Ländern auf, wo die Gesamtinzidenz etwa 1 von 20.000 Menschen beträgt. In der Verwaltungsregion Saguenay-Lac-Saint-Jean der Provinz Quebec erkrankt sogar 1 von 1846 Menschen an Tyrosinämie Typ 1.
Behandlung
Bei korrekter Erkennung und geeigneter medizinischer Versorgung wird erwartet, dass die meisten, wenn nicht sogar alle betroffenen Kinder ein Leben frei von Leber- und/oder Nierenerkrankungen führen können.
Die Therapie der Wahl bei Tyrosinämie Typ 1 ist Nitisinon in Kombination mit einer Tyrosin-/Phenylalanin-reduzierten Diät. Nitisinon, auch 2-[2-Nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedion (NTBC) genannt, ist ein potenter Hemmstoff der 4-Hydroxyphenylpyruvat-Dioxygenase (HPPD), die die Umwandlung von 4-Hydroxyphenylpyruvat in Homogentisinsäure katalysiert. Da Nitisinon die frühe Abbaukaskade von Tyrosin hemmt, minimiert es die Bildung von Fumarylacetoacetat und Maleylacetoacetat. Angesichts der Verfügbarkeit von NTBC ist die Option einer Lebertransplantation als HT-1-Behandlung auf die Fälle beschränkt, bei denen eine maligne Erkrankung oder eine dekompensierte, NTBC-resistente Lebererkrankung vorliegt.
Chinsky JM. Genet Med. 2017
Shaikh S et al. BJR Case Rep. 2018