SELTENE ERKRANKUNGEN
Gaucher-krankheit
Die Gaucher-Krankheit ist eine seltene, autosomal-rezessive, genetisch bedingte Störung. Sie wird durch einen Mangel des lysosomalen Enzyms Glukozerebrosidase (auch als saure β-Glucosidase bezeichnet) verursacht und führt zu einer Anreicherung des Substrats Glukosylzeramid. Dieser Prozess kann zu einer Splenomegalie und Hepatomegalie (Vergrößerung der Milz und Leber) und zu Knochenläsionen führen.
Geschichte der Erkrankung und ihrer medikamentösen Behandlung

Die Krankheit wird erstmals von dem Medizinstudenten Philippe Charles Ernest Gaucher (26.07.1854-25.01.1918) beschrieben, der sie bei einer 32-jährigen Frau mit vergrößerter Milz feststellte. Eine Post-mortem-Untersuchung ergab, dass die Zellen der Milz selbst vergrößert waren. Gaucher beschrieb diese klinischen und pathologischen Befunde in seiner Dissertation.
Dr. H. Lieb findet heraus, dass die chemische Ursache der Vergrößerung von Milz und Leber eine Anreicherung von Sphingoglykolipiden in den Zellen ist, die er irrtümlicherweise als Zerebroside bezeichnet. Die optische Drehung des wässerigen Spaltprodukts war jedoch mit dieser Schlussfolgerung nicht vereinbar.


Der französische Chemiker Dr. A. Aghion weist nach, dass die angereicherte Substanz Glukozerebroside sind.
Identifizierung von drei unterschiedlichen Phänotypen der Gaucher-Krankheit entsprechend dem Manifestationsalter der Symptomatik: Verlaufsform bei Erwachsenen ohne Schädigung des Zentralnervensystems (Typ 1), Verlaufsform bei Kindern mit akut neuronopathischer Beeinträchtigung (Typ 2) und juvenile oder chronisch neuronopathische Verlaufsform (Typ 3).

Dr. Roscoe Brady (11.10.1923–13.06.2016) und seine Gruppe zeigen, dass bei Menschen mit Gaucher-Krankheit das Lipid zwar normal gebildet wird, aber keine ausreichenden Mengen des Enzyms „Glukozerebrosidase“ für dessen Abbau und Elimination aus dem Körper. Der Befund, dass bei allen Patienten etwas von dem Enzym nachweisbar ist, erwies sich später beim Versuch ihrer Behandlung als äußerst wichtig. Dr. Brady ist ebenfalls für seine hervorragenden Leistungen bei der Identifizierung der Defekte in der Niemann-Pick-Krankheit, Fabry-Krankheit und Tay-Sachs-Krankheit bekannt.
Dr. Brady empfiehlt, die Gaucher-Krankheit durch Ersatz des fehlenden Enzyms zu behandeln (Enzymersatztherapie, ERT). Unter Verwendung menschlicher Plazenta isoliert Dr. Peter Pentchev aus dem Team von Dr. Brady eine winzige Probe gereinigter Glukozerebrosidase.
Dr. Bradys Team arbeitet einen Test auf Grundlage der Enzymaktivität aus, um eine potentielle Entwicklung der Gaucher-Krankheit zu erkennen, sowie ein pränatales Diagnoseverfahren.
Zwei Patienten erhalten das gereinigte Enzym Glukozerebrosidase durch intravenöse Injektion. Aufgrund der guten biochemischen Ergebnisse entschließt sich Dr. Brady ein Verfahren zu entwickeln, um größere Mengen des Enzyms zu gewinnen und damit weitere klinische Studien durchzuführen.
Ein großtechnisches Reinigungsverfahren wird fertiggestellt, aber diese Aufbereitung des Enzyms führt in den klinischen Studien zu uneinheitlichen Ergebnissen aufgrund des unzureichenden Enzym-Transports zu den Makrophagen, den hauptsächlich betroffenen Zielzellen.
Dr. Shoji Tsuji und sein Team entdecken die erste Genmutation als Ursache der Gaucher-Krankheit, die sich somit als autosomal-rezessive Erbkrankheit erweist. Das bedeutet, dass beide Glukozerebrosidase-Gene, die ein Individuum erbt – eines von der Mutter und eines vom Vater –, mutiert sein müssen, damit sich die Krankheit bei diesem Individuum entwickelt. Dr. Shoji Tsuji wird 2015 mit der Medaille für wissenschaftliche Errungenschaften in der Neurologie für seine großen Leistungen bei der Aufklärung der Gaucher-Krankheit ausgezeichnet.


Dr. Norman Radin postuliert einen neuen Therapieansatz, mit dem die Glukozerebrosid-Anreicherung durch Hemmung des Syntheseweges verringert werden soll. Dieser potentielle Therapieansatz wird als Substratreduktionstherapie (SRT) bezeichnet.
Ein 1984 untersuchtes Verfahren wird optimiert. Es verbessert maßgeblich den Transport der Glukozerebrosidase zu den Makrophagen und führt zu neuen klinischen Studien an Patienten mit Gaucher-Krankheit Typ 1. Die erzielten Ergebnisse belegen einen signifikanten klinischen Nutzen zusammen mit einer ausgeprägten Größenreduktion der Leber und Milz und einer Verringerung der Knochenschäden. Ein einschränkender Faktor ist jedoch, dass eine große Menge Enzym pro kg Körpergewicht erforderlich ist.
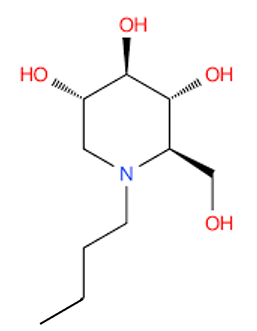
Die Identifizierung des Glukose-Analogons N-Butyl-desoxynojirimycin (NB-DNJ, Miglustat), eines spezifischen Hemmstoffs des Zeramid-spezifischen Enzyms Glukosyltransferase (CSG), das am ersten Schritt der Glykosphingolipid-Synthese beteiligt ist, einschließlich Glukozerebrosid. NB-DNJ wurde anfänglich als antiviraler Wirkstoff gegen bestimmte Krankheiten, u. a. HIV, entwickelt.
Neue klinische Studien zur ERT bei Patienten mit Gaucher-Krankheit Typ 2 und Typ 3 zeigen eine bemerkenswerte Verbesserung bei der Behandlung systemischer Krankheitssymptome, aber die Wirksamkeit der Behandlung in Bezug auf die neurologischen Symptome bleibt wegen der schwierigen Passage des Enzyms durch die Blut-Hirn-Schranke problematisch. Das ist ein stark limitierender Faktor der ERT, der zusammen mit den damit verbundenen hohen Kosten die Behandlungsmöglichkeit selbst handlungsbedürftigster Patienten beschränkt.
Die einjährige offene Studie zur Prüfung der Wirkung von Miglustat bei der Gaucher-Krankheit Typ 1 wird abgeschlossen. Die Studienergebnisse zeigen bei Leber und Milz eine 19%ige Volumenreduktion.
Die EMEA erteilt dem Schweizer Unternehmen Actelion Pharmaceuticals die Marktzulassung für Zavesca® (Miglustat 100 mg Kapseln), die erste orale Therapie für die Gaucher-Krankheit Typ 1. Miglustat wirkt als SRT, indem es die Bildung von Glukozerebrosiden verhindert. Zavesca® ist eine Behandlungsoption für Patienten, bei denen eine Enzymersatztherapie aus diversen Gründen wie Allergie und Hypersensitivität nicht angezeigt ist.
Zavesca® wird von der US-FDA zugelassen.
Eine offene, multizentrische Phase-III-Studie zur Prüfung der Langzeit-Sicherheit und ‑Wirksamkeit von Zavesca® bei Erwachsenen mit Gaucher-Krankheit Typ 1 wird abgeschlossen. Die Ergebnisse zeigen, dass eine Behandlung mit Miglustat es ermöglicht, die Lebergröße insgesamt konstant zu halten.
Symptomatik
Bei der Gaucher-Krankheit kommt es zu Hepatosplenomegalie (gleichzeitige Vergrößerung von Leber und Milz), Zytopenie, mitunter zu schwerwiegender Knochenbeteiligung und bei bestimmten Verlaufsformen zu neurologischen Schädigungen.
Die häufigste Form der Krankheit (Gaucher-Krankheit Typ 1), bei der es in der Regel zu keinen neurologischen Schädigungen kommt, hat sehr unterschiedliche klinische Verlaufsformen, die von lebenslang asymptomatisch bis zu Manifestation im jungen Kindesalter reichen.
Bei mehr als 90 % der Patienten wird eine mitunter extreme Splenomegalie mit einem Milzgewicht bis zu mehreren Kilogramm mit Bauchschmerzen und aufgeblähtem Bauch beobachtet. Tatsächlich kann dies das einzige klinische Anzeichen sein. Bei einer Milzvergrößerung ist das Risiko einer Milzruptur erhöht, zu der es aber nur sehr selten kommt.
Bei 60 %–80 % der Patienten wird eine Hepatomegalie beobachtet. Es kann auch zu einer Leberruptur kommen, die sich in heftigen Schmerzen im Bauchraum äußert.
Bei einer Knochenbeteiligung kommt es zu akuten Schmerzen in Form sehr schmerzhafter Schübe, sog. Knochenkrisen, vorwiegend im Bereich des Beckens und unteren Gliedmaßen (seltener der oberen Gliedmaßen), und/oder zu chronischen Schmerzen.
Inzidenz
In der allgemeinen Population beträgt die Inzidenz etwa 1/60.000 bis 1/40.000 Geburten und bis zu 1/800 in der ethnischen Population der aschkenasischen Juden.
Behandlung
Das Ziel ist die Behandlung der Patienten, bevor es zu Komplikationen kommt, deren Folgen irreversibel sind und zu Behinderungen führen, wie massive Vergrößerung und Fibrose der Milz, Gefäßnekrose, sekundäre Osteoarthritis, Wirbelsäulenkompression und Wirbelkompressionsfrakturen, Leberfibrose und Lungenfibrose.
Für die Gaucher-Krankheit sind gegenwärtig zwei spezifische Behandlungsoptionen verfügbar: die Enzymersatztherapie (ERT) für die Zufuhr der in den Zellen fehlenden Glukozerebrosidase und die Substratreduktionstherapie (SRT) zur Reduktion des in den Zellen gespeicherten Glukosylzeramids durch dessen verringerte Produktion. Miglustat hemmt die Glukosylzeramidsynthase und verringert dadurch die Glukosylzeramid-Synthese.
Stirnemann J et al. Int J Mol Sci. 2017